Excited vs. Hot Ground State Reactivity in the Photochemistry of Alkylcyclobutenes in the Gas Phase and Solution
William J. Leigh and Bruce H.O. Cook
Department of Chemistry, McMaster University, 1280 Main Street West, Hamilton, ON Canada L8S 4M1
![]()
INTRODUCTION
Direct irradiation of alkylcyclobutenes in solution results in competing ring opening to the isomeric dienes and cycloreversion to yield an alkyne and alkene. Ring opening is non-stereospecific, while formation of alkene via cycloreversion is stereospecific. For example, Equation 1 shows the products obtained from 214-nm photolysis of cis- and trans-1,2,3,4-tetramethylcyclobutene in pentane solution. The relative yields of each of the products are indicated:
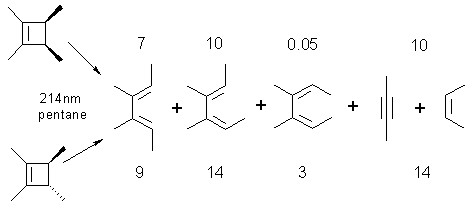
Leigh, W.J.; Zheng, K.; Clark, K.B. Can. J. Chem. 1990,
68, 1988.
Leigh, W.J.; Postigo, J.A. J. Am. Chem. Soc. 1994, 117, 1688.
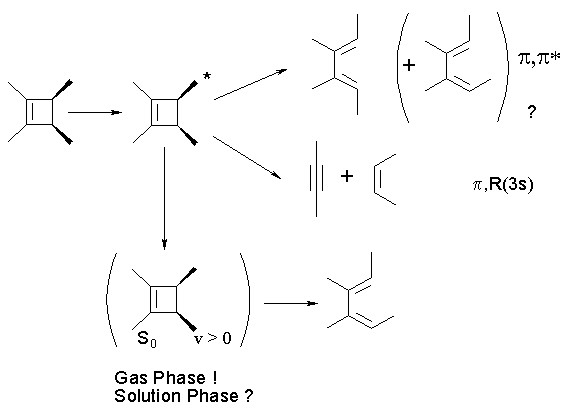
The two types of products have been explained in terms of two distinct mechanisms arising from different excited states. Ring opening is thought to occur in the p,p* excited singlet state to produce the formally photochemically-allowed diene isomer as the initial product, but proceeds adiabatically to form the excited diene which decays to form a mixture of diene isomers. Cycloreversion is believed to proceed mainly from an excited singlet state which in solution appears in roughly the same spectral position as the gas phase p,R(3s) Rydberg absorption. Both the Rydberg state (in the gas phase) and the "Rydberg-like" state (in solution) are at lower energies than the p,p* excited singlet state, at least in 1,2-dialkylcyclobutenes.
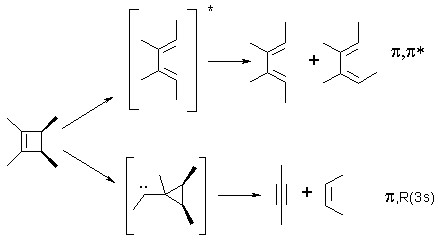
The ring opening mechanism has been experimentally investigated quite thoroughly by our group. One of the best pieces of evidence in support of the adiabatic mechanism came from a study where the ratio of diene isomers formed upon irradiation is compared to that formed upon irradiation of the formally allowed diene isomer. Note that this particular diene is constrained to be in the s-cis conformation, the same as should be produced (initially) in the electrocyclic ring opening of cyclobutene:
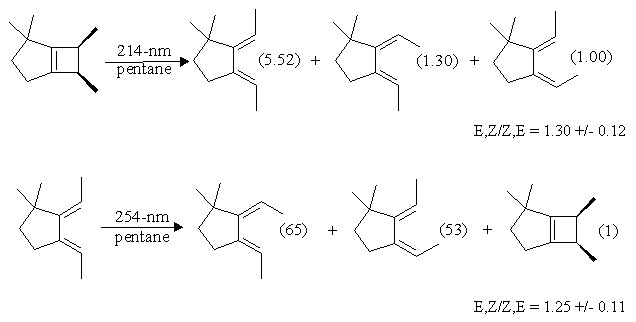
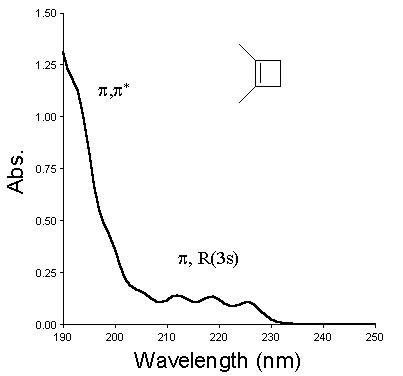
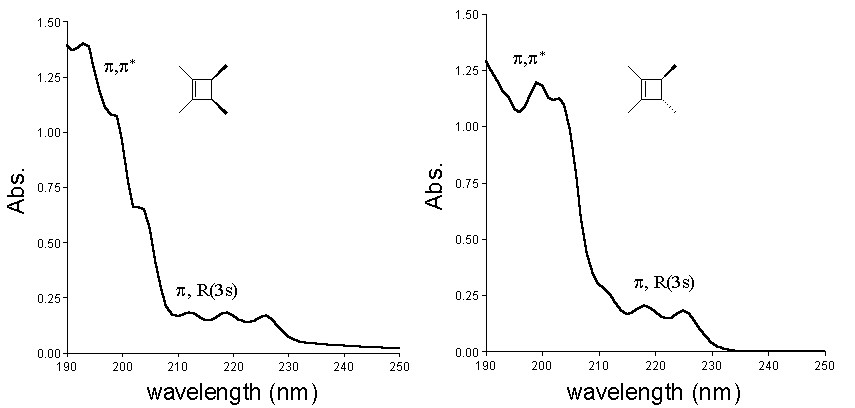
We have continued to worry about the involvement of the "Rydberg-like" excited singlet state in solution phase cyclobutene photochemistry. While the photochemistry of aliphatic alkenes in solution has often been explained in terms of the involvement of the p,R(3s) Rydberg excited state, there remain serious questions as to whether pure Rydberg states can exist in condensed phases. Solution phase alkene photochemistry can just as conveniently be explained as due to the population of a valence (p,s*) state, which either underlies or is mixed with the Rydberg state in the gas phase. In order to probe the true involvement of this excited state in solution phase alkylcyclobutene photochemistry and compare its behavior to the gas phase p,R(3s) state, we have carried out a comparison of the gas and solution phase photochemistry of three compounds: 1,2-dimethylcyclobutene (DMCB) and cis- and trans-1,2,3,4-tetramethylcyclobutene (c- and t-TMCB, respectively). The gas phase uv absorption spectra (1 atm, SF6) are shown below, with the p,p* and p,R(3s) absorptions assigned. The solution phase spectra lack the fine structure observed in the gas phase spectra, but tail out to similar wavelengths.
W.J. Leigh, K.C. Zheng, and K.B. Clark, Can. J. Chem. 68, 1988 (1990).
These spectra suggest that by irradiating at 214-nm or (especially) 228-nm, it should be possible to selectively populate the "pure" Rydberg state in the gas phase, and compare its behavior to the photochemistry observed upon irradiation at the same wavelengths in solution.
RESULTS
Photolyses were carried out in the gas phase (~250 torr cyclobutene diluted to 1 atm with SF6) and in deoxygenated isooctane or cyclohexane solution (0.05-M) using 193, 214, and 228-nm light sources. The data obtained are summarized as product ratios (ring opening / cycloreversion; EE:EZ:ZZ diene distributions; photochemically allowed / thermally allowed diene isomers) in Table 1. The table also contains the photostationary state (pss) ratios for the dimethylhexadienes formed from c- and t-TMCB, determined in both the gas phase and solution with 228-nm excitation.
Table 1. Product ratios from photolysis of DMCB, c-TMCB and t-TMCB in the gas phase and in hydrocarbon solution (25 oC). "RO/CR" refers to the ratio of ring-opening (all isomers) to cycloreversion products, "EE:EZ:ZZ" is the distribution of the three isomeric dienes from c- and t-TMCB, and "Therm/Photo" is the ratio of thermally-allowed to photochemically-allowed diene isomers from c- and t-TMCB.
Gas Phase |
Hydrocarbon Solution |
Cmpd. |
l ex (nm) |
RO/CR |
EE:EZ:ZZ |
Therm / Photo |
RO/CR |
EE:EZ:ZZ |
Therm / Photo |
193 |
3.0 |
- |
- |
1.3 |
- |
- |
||
| DMCB | 214 |
2.4 |
- |
- |
0.92 |
- |
- |
|
228 |
2.0 |
- |
- |
0.79 |
- |
- |
193 |
1.6 |
1 : 2 : 0 |
2.0 |
1.9 |
1.6 : 1 : 0 |
0.63 |
||
| c-TMCB | 214 |
1.0 |
1 : 6 : 0 |
6.0 |
1.1 |
1 : 1.5 : 0 |
1.5 |
|
228 |
0.82 |
<1 : 99 : <1 |
90 |
0.69 |
1 : 5.2 : 0 |
5.2 |
193 |
1.6 |
6 : 2 : 1 |
4.0 |
1.5 |
5.2 : 8.5 : 1 |
0.74 |
||
| t-TMCB | 214 |
1.3 |
3.8 : 1 : 1 |
4.9 |
1.0 |
2 : 4.1 : 1 |
0.74 |
|
228 |
0.74 |
2.8 : 1.5 : 1 |
2.5 |
0.32 |
1 : 4.3 : 2.7 |
0.85 |
pss |
228 |
- |
1 : 7 : 2 |
- |
- |
1 : 7.1 : 4.4 |
- |
The following conclusions can be made from the data:
| the yields of ring opening relative to cycloreversion products are similar or higher in the gas phase than in solution | |
| the yields of ring opening relative to cycloreversion products decrease with increasing excitation wavelength in both the gas and solution phases |
| irradiation of c-TMCB at 228-nm produces mainly E,Z-3,4-dimethyl-2,4-hexadiene, the thermally-allowed diene isomer, in both the gas phase and solution. In the gas phase, this isomer is formed with >95% stereospecificity! | |
| irradiation of t-TMCB at 228-nm produces all three diene isomers. In solution, the diene distribution is very close to the diene pss, suggesting that secondary diene photolysis is skewing the results in this case. Note that in the gas phase, the thermally-allowed (EE and ZZ) diene isomers are again the major ring opening products. |
Future Research
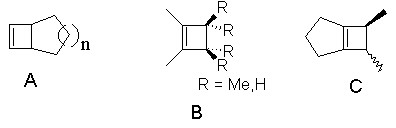
Further investigations of the extent of the contribution of this new mechanism to cyclobutene ring opening are in progress. These include study of:
| gas and solution phase 228-nm photochemistry of bicyclic cyclobutenes (A) whose thermal ring opening proceed with higher activation energies than those studied here. | |
| steric effects on long wavelength (monocyclic) cyclobutene ring opening (compounds B; see Leigh & Postigo, JACS 117, 1688 (1995)). | |
| gas and solution phase 228-nm photochemistry of bicyclic cyclobutenes (C) whose behavior are consistent with the adiabatic p,p* ring opening mechanism. |
Acknowledgements
Dr. William J. Leigh Nick Toltl Ed Lathioor
Tracy Morkin Xien Fu Zhang Tom Owens