Photochemistry of Phthalimides: Decarboxylation, Addition, Macrocyclization and Deprotection [1]
Michael Oelgemöller* and A. G. Griesbeck†
* Inoue Photochirogenesis Project, ERATO-JST, 4-6-3 Kamishinden, Toyonaka-shi, Osaka 560-0085, Japan; oelge@inoue.jst.go.jp
† Institute of Organic Chemistry, University of Cologne, Greinstr. 4, D-50939 Köln, Germany
Due to their favorable photophysical properties [2] the photochemistry of phthalimides have been intensively studied by us [3] and others [4] over the last decades with respect to applications in synthetic organic photochemistry. Especially interesting compounds are N-phthaloyl amino acids, which offer a broad spectrum of photochemical reactions including homolytic CH-activation and photoinduced electron transfer (PET). The latter case has been investigated by us either for sulfur-[5] or carboxylate-substituted [6] derivatives.
1. Photodecarboxylative addition of carboxylates to phthalimides
C-unprotected a-amino acid derivatives solely underwent a-decarboxylation when irradiated in several organic solvents [7]. Remote carboxylic groups were photochemically inert but were easily activated via transformation into the corresponding potassium salts and irradiation in acetone/water mixtures [6]. Medium and large ring systems were available using this method, and many functional groups were tolerated in the spacer chain. Thus, also an intermolecular version of this reaction became in principle feasible, and was studied in detail.
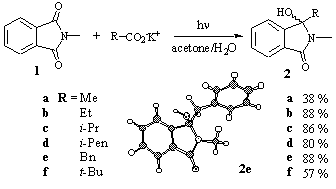
In most cases, N-methyl phthalimide 1 was used as a model substrate and the carboxylate compound was varied. From unfunctionalized potassium alkyl carboxylates, the corresponding hydroxy phthalimidines 2a-f were obtained in moderate to good yields between 38-88% (Scheme 1) [8]. In all cases, the carboxylate salt was used in a 2-5 molar excess in order to drive the reaction to completion.
Scheme 1
As a side reaction, the formation of “simple” decarboxylation products (CO2-/H-exchange) was competing with the addition reaction, and for 1-adamantane carboxylate, however, the corresponding hydrocarbon adamantane became the sole product whereas N-methyl phthalimide could be reisolated almost quantitatively [8].
As a model reaction for a large scale preparation (using a 308 nm XeCl excimer light source), the photodecarboxylative addition of potassium iso-butyrate to 1 was performed (Scheme 2). This reaction proceeded smoothly in a 3:1 (vol.-%) water-acetone mixture using 6 equivalents of the alkyl carboxylate and a 0.02 M solution of 1. The reaction progress was monitored by continuous pH control and gas chromatographic determination of the substrate/product ratio [9].
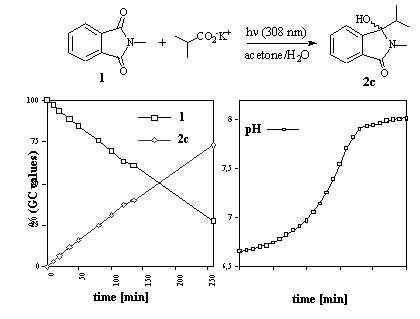

Scheme 2
In contrast to the (thermal) Grignard addition, other carbonyl groups, which were present in the substrates, did not react with the intermediary produced alkyl radicals. This was demonstrated for the methyl esters of N-phthaloyl amino acids 3a-i [8]. With these substrates, the photoinduced alkylation occurred regioselectively at the imide and not at the ester carbonyl group (Scheme 3). Unfortunately, the diastereoselectivity was only negligible to moderate [10].
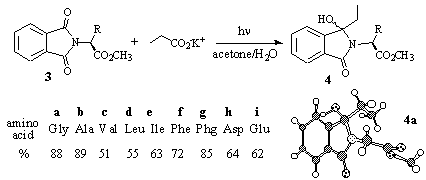
Scheme 3
The presence of a heteroatom in the carboxylate chain either strongly increased or decreased the addition efficiency [11]. a-Thioalkyl and a-oxoalkyl-substituted carboxylates led to the corresponding reduction products in moderate to good yields (57-90%) [12]. In contrast to that, the b-thioalkyl-substituted carboxylate was inert under the reaction conditions, whereas the corresponding b-oxoalkyl carboxylate reacted efficiently to give the addition product 5g in 51% yield (Scheme 4).
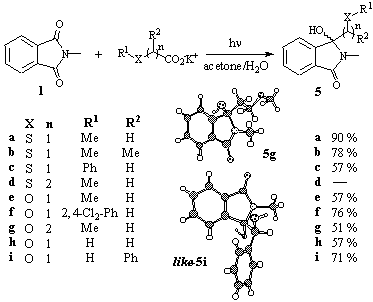
Scheme 4
From the comparison of the sulfur- and the oxygen-substituted carboxylates, oxidation of the heteroatom is assumed to play the dominant role in the sulfur case. The resulting sulfur radical cation destabilizes the terminal a-C-C-bond and accelerates CO2-extrusion. Dialkylethers are much less readily oxidized and instead, oxidation of the carboxyl group occurs. This assumption is in accord with the oxidation potentials of the competing donor groups, which increase for R2S < RCO2- < R2O < ROH. In case of the b-thio substrate, the sulfur atom acts as a “hole trap” (fast non-productive back electron transfer) and prevents oxidation of the carboxylate.
a-Keto carboxylates are important substrates for the enzymatic synthesis of a-amino acids [13]. Additionally, they are highly photoactive substrates, and sensitive towards decarboxylation when irradiated [14]. This class of carboxylic acids was therefore chosen for further functionallization in the photodecarboxylative addition reaction to phthalimides. When using glyoxylate 6a as well as the secondary or tertiary a-keto carboxylates 6b-d, respectively, the phthalimidines 7a-d were obtained in acceptable to high yields between 52-86% (Scheme 5) [8]. Obviously, photodecarboxylation is rapidly followed by photodecarbonylation for theses substrates.
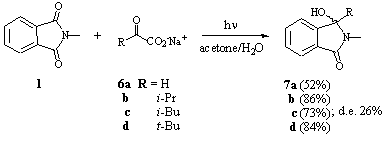
Scheme 5
The primary a-keto carboxylates 6e and f, however, solely gave acylation instead of alkylation products (Scheme 6) [15]. The primary acylation product 9 was isolated for the a-keto leucine 6e beside ca. equal amounts of a second photoproduct. The structure of this additional product could not be solved until pyruvate 6f was used as the reactant. 6f solely yielded the structurally related product, which was identified by X-ray structure analysis as the dihydroisoquinolinyl ester 8b. Obviously, two equivalents of a-keto carboxylates were involved in the addition reaction to give 8.
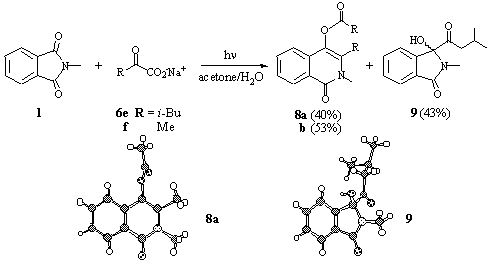
Scheme 6
The formation of 8 can not be explained by addition of an acyl radical formed by decarboxylation of an excited keto carboxylate 6 because the anion does not undergo efficient photodecarboxylation [16]. Thus, the second decarboxylative addition step can be induced either thermally by acylation of an intermediary acyliminium cation [17] or photochemically involving the excited triplet state of the postulated isoquinoline intermediate. Its parent compound phthalonimide indeed shows a remarkable photochemical reactivity [18].
2. PET-induced macrocyclizations
The combination of the phthalimide chromophore and the thioether and carboxylate functionalities in the same molecule leads to acceptor-donor-donor triades. Due to its lower lying oxidation potential the thioether fragment is expected to be the more reactive donor for photoinduced electron transfer than the carboxylate. This behavior was already observed for the intermolecular case (vide infra) [11]. In agreement, the potassium salts of 10 showed a remarkable shift in the efficiency of cyclization (Scheme 7) [19, 20]. This shift corresponds to the change in the carbon spacer chain between the thioether and the carboxylate functionality (n). In contrast, the number of carbons between the phthalimide chromophore and the sulfur atom (m) does not influence the cyclization efficiency at all, and the cyclization proceeds regioselectively at the terminal carbon-position. Ring closure at the “inner” CH2-group in a-position to sulfur (as described by Sato and Kanaoka for the methyl thioethers [21]) was not observed.
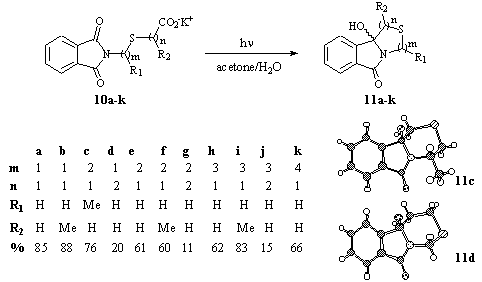
Scheme 7
The previously assumed intramolecular photoinduced electron transfer mechanism from the thioether to the triplett excited phthalimide [21] was found to operate only for direct excitation of the chromophore [22]. Under the acetone sensitized conditions described in here, intermolecular electron transfer to the triplett excited acetone is postulated [22]. In contrast to the thiomethyl systems [21], no proton rearrangement but elimination of the carboxylate occurs for the a-carboxylate derivatives. This corresponds nicely to the a-stannyl- and a-silylthiomethoxy derivatives developed by Ikeno [23] and Yoon and Mariano [24]. The latter authors have demonstrated for N-(aminoethyl) phthalimides carrying CH2-LG as terminal group that quantum efficiencies increase with LG = H < TMS < CO2-, respectively [25]. The presence of the appropriate leaving group CO2 enhances the chemical reactivity of the radical ion pair and reduces the non-productive back electron transfer (BET) reaction. For the b-carboxylate systems product formation can no longer be explained by electron transfer from the sulfur, and subsequent decarboxylative cyclization. In these cases and in analogy to the phthalimido w-carboxylic acids [6] “productive” electron transfer from the carboxylate seems to by the pathway of choice. The remarkable drop in the efficiency can be interpreted by the weaker donor properties of the carboxylate. The low quantum and chemical yields for cyclization (compared to the a-carboxylate derivatives), and the high amounts of reisolated unreactive starting material (54-76%) support that the primary electron transfer from the thioether function is followed by rapid BET. The higher amounts of unselective fragmentations (as detected by GC-MS) also indicate that side reactions from the sulfur radical cation can compete with the decarboxylative cyclization.
Other interesting classes of substrates for PET-induced macrocyclizations [26] are the C-protected MTM- and MTE-esters of phthaloyl amino acids, which allow the investigation of the conformational restricted ester-linker fragment [27]. Although this concept has already been realized for macrocyclization reactions using ester [28] as well as peptide [29] linked substrates and the thiomethyl group as electron donor, only little was known about the relationship between cyclization efficiency and the linker structure, especially for precursors to medium-sized ring systems. We have recently demonstrated for the decarboxylative cyclization of N-phthaloyl peptides [30] and esters [31] that rigid spacer groups and spacers with H-bond activity can strongly influence the efficiency for the photochemical macrocyclizations. The photolyses of the MTM-ester derivatives 12 resulted in two groups of products: cyclization and "deprotection".
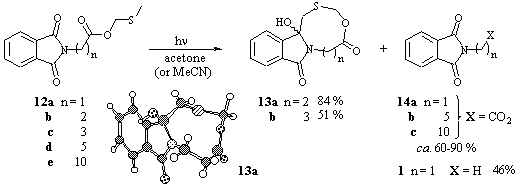
Scheme 8
Substrates 12b and c gave the corresponding macrocycles 13a and b in 84 and 51% yield, respectively (Scheme 8) [32]. On the contrary, the esters 12a, d and e mainly yield the free N-phthaloyl amino acids 14a-c. Only the glycine compound 12a was further decarboxylated to give 46% of N-methyl phthalimide 1 in acetone (55% conversion), whereas photolysis in acetonitrile resulted in a 2:1 mixture of N-phthaloyl glycine 14a and 1.
Only cyclization was observed for the less rigid MTE-esters 15a and b (Scheme 9). However, for 15b the corresponding olefinic dehydration product E-17 was obtained in 72% yield. The Z-isomer was identified in minor amounts (E:Z ca. 85:15) in the proton NMR of the crude product mixture, but could not be isolated. The structures of 16 and E-17 were solved by X-ray structure analysis.
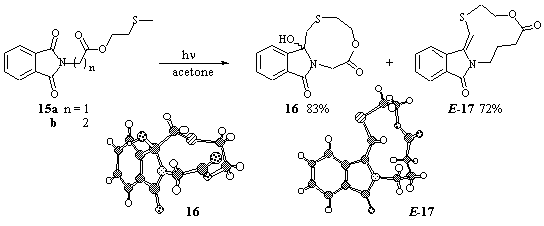
Scheme 9
As the key-step in the reaction mechanism intermolecular photoinduced electron transfer is postulated from the thioether to the triplet excited sensitizer (acetone). Proton transfer from the terminal a-position of the sulfur and C-C bond formation leads to the cyclization products 13 and 15. The olefinic compound 16 is formed after hydrolysis, a process that is known to proceed in the presence of catalytic amounts of acids [3]. For some of the MTM-esters, however, deprotection to the free acids 14 is favored, but the final product of the MTM-group is not clear at present [33]. In these special cases the MTM-group acts like a photoremovable protecting group (PRPG) [34]. For the glycine compound 12a deprotection is followed by rapid photochemical a-decarboxylation [7]. The (compared to the MTE-esters) prolonged irradiation time for the MTM-esters rationalizes the assumption that higher flexibility favors cyclization, whereas lower flexibility favors deactivation processes like BET or deprotection.
In order to study the diastereoselectivity of the radical combination step, the two model systems 18a and b were examined. These compounds differ only in the relative position of the linking ester group along the spacer chain (Scheme 10). Irradiation of 18a gave diastereoselectively and in 61% yield cis-19a. Again, prolonged irradiation was necessary to run the reaction to 100% conversion. In contrast, the MTE-ester 18b was readily transferred into the corresponding macrocyclic mixture 19b after relatively short irradiation time, and the diastereoisomers cis- and trans-19b were isolated in a ca. 4:1 ratio and in 88% yield. According to the conformational conditions inside the molecule, the additional carbon in the methylthioalkyl fragment in 18b increases the tendency for cyclization on the one hand, but lowers the stereoselectivity on the other hand.
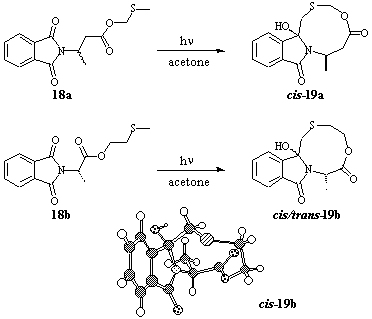
Scheme 10
As a conclusion, a large number of useful photochemical transformations in the phthalimide series were developed. These processes allow the photochemical synthesis of interesting new compounds via addition, ring expansion, cyclization or fragmentation reactions. Beside the spacer fragments shown in here also peptide or olefinic linkers were examined [6], and next to phthalimides other imides like quinolinic- and trimellitic acid imides [35] were suitable chromophores for PET-reactions.
References and Notes:
[1] a) M. Oelgemöller EPA Newsletter 2000, 69, 11.
b) M. Oelgemöller EPA Newsletter 1999, 66, 63.
[2] a) A. G. Griesbeck, H. Görner J. Photochem. Photobiol. A: Chem. 1999, 129, 111.
b) T. C. Barros, S. Brochsztain, V. G. Toscano, P. Berci Filho, M. J. Politi J. Photochem. Photobiol. A: Chem. 1997, 111, 97.
c) V. Witgens, P. Valet, J. Kossanyi, L. Biczok, A. Demeter, T. Berces J. Chem. Soc., Faraday Trans. 1994, 90, 411.
d) J. D. Coyle, A. Harriman, G. L. Newport J. Chem. Soc., Perkin Trans. 2 1979, 799.
[3] a) A. G. Griesbeck Chimia 1998, 52, 272.
b) A. G. Griesbeck EPA Newsletter 1998, 62, 3.
c) A. G. Griesbeck Liebigs Ann. 1996, 1951.
[4] a) H. Mauder, A. G. Griesbeck CRC Handbook of Organic Photochemistry and Photobiology, W. M. Hoorspool and P.-S. Song (Eds.), CRC Press: Boca Raton, 1994, pp. 513.
b) J. D. Coyle Synthetic Organic Photochemistry, W. M. Hoorspool (Ed.), Plenum Press: New York, 1984, pp. 259.
c) P. H. Mazzocchi Org. Photochem. 1981, 5, 421.
d) Y. Kanaoka Acc. Chem. Res. 1978, 87, 407.
e) Y. Kanaoka J. Soc. Org. Synth. Chem. (Tokyo) 1975, 33, 949 (in Japanese).
[5] a) A. G. Griesbeck, J. Hirt, W. Kramer, P. Dallakian Tetrahedron 1998, 54, 3169.
b) A. G. Griesbeck, J. Hirt, K. Peters, E.-M. Peters, H. G. von Schnering Chem. Eur. J. 1996, 2, 1388.
[6] a) A. G. Griesbeck, W. Kramer, M. Oelgemöller Synlett 1999, 1169.
b) W. Kramer, A. G. Griesbeck, F. Nerowski, M. Oelgemöller J. Inf. Rec. 1998, 24, 81.
c) A. G. Griesbeck, A. Henz, W. Kramer, J. Lex, F. Nerowski, M. Oelgemöller, K. Peters, E.-M. Peters Helv. Chim. Acta 1997, 80, 912.
d) A. G. Griesbeck, A. Henz, K. Peters, E.-M. Peters, H. G. von Schnering Angew. Chem. 1995, 107, 498; Angew. Chem. Int. Ed. Engl. 1995, 34, 474.
[7] a) Y. Takahashi, T. Miyashi, U. C. Yoon, S. W. Oh, J. C. Sung, Y.-J. Lee, M. Mancheno, Z. Su, D. F. Falvey, P. S. Mariano J. Am. Chem. Soc. 1999, 121, 3926.
b) A. G. Griesbeck, A. Henz Synlett 1994, 931.
c) Y. Sato, H. Nakai, T. Mizoguchi, M. Kawanishi, Y. Hatanaka, Y. Kanaoka Chem. Pharm. Bull. 1982, 30, 1263.
[8] A. G. Griesbeck, M. Oelgemöller Synlett 1999, 492.
[9] A. G. Griesbeck, W. Kramer, M. Oelgemöller Green Chemistry 1999, 1, 205.
[10] The degree of diastereoselectivity can be explained by a sterical 1, 3-induction from the substituent of the amino acid. X-ray structure of 3b, see: K. Peters, E.-M. Peters, A. G. Griesbeck, M. Oelgemöller, A. Bartoschek Z. Kristall. NCS 1999, 214, 107.
[11] A. G. Griesbeck, M. Oelgemöller Synlett 2000, 71.
[12] X-ray structure of unlike-5b, see: K. Peters, E.-M. Peters, M. Oelgemöller, J.-M. Cho, A. G. Griesbeck Z. Kristall. NCS 2000, 215, 37.
[13] a) K. Drauz, H. Waldmann Enzyme Catalysis in Organic Synthesis, Vol. 2, VCH: Weinheim, 1995.
b) G. Krüger Methods Org. Chem. (Houben-Weyl), E 16d, part I, D. Klamann (Ed.), 1992.
[14] a) H. Görner, L. J. Currell, H. J. Kuhn J. Phys. Chem. 1991, 95, 5518.
b) H. J. Kuhn, H. Görner J. Phys. Chem. 1988, 92, 6208.
c) R. S. Davidson, D. Goodwin J. Chem. Soc. Perkin Trans. II 1982, 1559.
d) R. S. Davidson, D. Goodwin, P. Fornier de Violet Chem. Phys. Lett. 1981, 78, 471.
[15] A. G. Griesbeck, M. Oelgemöller, J. Lex Synlett 2000, 1455.
[16] a) A. Defoin, R. Defoin-Straatmann, K. Hildenbrand, E. Bittersmann, D. Kreft, H. J. Kuhn J. Photochem. 1986, 33, 237.
b) P. A. Leermarkers, G. F. Vesley J. Am. Chem. Soc. 1963, 85, 3776.
c) P. A. Leermarkers, G. F. Vesley J. Org. Chem. 1963, 28, 1160.
[17] a) H. De Koning, W. N. Speckamp Methods Org. Chem. (Houben-Weyl), 21b, 1995, pp. 1953.
b) H. Hiemstra, W. N. Speckamp Comprehensive Organic Synthesis, Vol. 2, C. H. Heathcock (Ed.), Pergamon Press: Oxford, 1991, pp. 1047.
[18] a) R. Suau, E. P. de Inestrosa Villatoro Tetrahedron 1995, 51, 6293.
b) R. Suau, E. P. de Inestrosa Villatoro Tetrahedron 1994, 50, 4987.
[19] A. G. Griesbeck, M. Oelgemöller, J. Lex, A. Haeuseler, M. Schmittel Eur. J. Org. Chem. 2000, submitted.
[20] X-ray structure of 11g, see: K. Peters, E.-M. Peters, A. G. Griesbeck, M. Oelgemöller, Z. Kristall. NCS 1998, 213, 757.
[21] a) Y. Hatanaka, Y. Sato, H. Nakai, M. Wada, T. Mizoguchi, Y. Kanaoka Liebigs Ann. Chem. 1992, 1113.
b) Y. Sato, H. Nakai, M. Wada, T. Mizoguchi, Y. Hatanaka, Y. Migita, Y. Kanaoka Liebigs Ann. Chem. 1985, 1099.
[22] H. Görner, A. G. Griesbeck, T. Heinrich, W. Kramer, M. Oelgemöller Chem. Eur. J. 2000, in print.
[23] T. Ikeno, M. Harada, N. Arai, K. Narasaka Chem. Lett. 1997, 169.
[24] a) U. C. Yoon, J. W. Kim, J. Y. Ryu, S. J. Cho, S. W. Oh, P. S. Mariano J. Photochem. Photobiol. A: Chem. 1997, 106, 145.
b) U. C. Yoon, S. J. Lee, K. J. Lee, S. J. Cho, C. W. Lee, P. S. Mariano Bull. Korean Chem. Soc. 1994, 15, 154.
[25] a) Z. Su, P. S. Mariano, D. E. Falvey, U. C. Yoon, S. W. Oh J. Am. Chem. Soc. 1998, 120, 10676.
b) Z. Su, D. E. Falvey, U. C. Yoon, P. S. Mariano J. Am. Chem. Soc. 1997, 119, 5261.
[26] A. G. Griesbeck, A. Henz, J. Hirt Synthesis 1996, 1261.
[27] A. G. Griesbeck, M. Oelgemöller, J. Lex J. Org. Chem. 2000, in print.
[28] a) M. Wada, H. Nakai, K. Aoe, K. Kotera, Y. Sato, Y. Hatanaka, Y. Kanaoka Tetrahedron 1983, 39, 1273.
b) M. Wada, H. Nakai, K. Aoe, K. Kotera, Y. Sato, Y. Kanaoka Chem. Pharm. Bull. 1982, 30, 2275.
[29] Y. Sato, H. Nakai, M. Wada, T. Mizoguchi, Y. Hatanaka, Y. Kanaoka Chem. Pharm. Bull. 1992, 40, 3174.
[30] A. Bartoschek, A. G. Griesbeck, M. Oelgemöller J. Inf. Rec. 2000, in print.
[31] A. G. Griesbeck, F. Nerowski, J. Lex J. Org. Chem. 1999, 64, 5213.
[32] M. Oelgemöller, A. G. Griesbeck, W. Kramer, F. Nerowski J. Inf. Rec. 1998, 24, 87.
[33] For an analogue electrochemical deprotection, see: L. A. Barnhurst, Y. Wan, A. G. Kutateladze Org. Lett. 2000, 2, 799.
[34] For a review on PRPGs, see: V. N. R. Pillai Org. Photochem. 1987, 9, 225.
[35] A. G. Griesbeck, M. S. Gudipati, J. Hirt, J. Lex, M. Oelgemöller, H. Schmickler, F. Schouren J. Org. Chem. 2000, 65, 7151.