| Site home page | Conference home page | Discussion |
TRIPLET LIFETIME DEPENDENCE ON VIBRATIONAL ENERGY FOR GAS-PHASE
CARBONYL AROMATIC COMPOUNDS: BENZOPHENONE AND FLUORENONE
Institute of Molecular and Atomic Physics, National Academy of Sciences of Belarus,
Scaryna Av. 70, 220072 Minsk, Belarus
zalesskaya@imaph.bas-net.by
A strong increase in triplet decay rates by over three orders of magnitude under IR laser excitation of gas phase triplet molecules was revealed. The dependences of intensities and decay rates of CO2 laser induced delayed fluorescence on vibrational energy and bath gas pressure were used to study dominant relaxation pathways in the lowest triplet state T1 within a wide range of vibrational energy Evib. With this end in view, the excess energy dependence for decay rates of all nonradiative relaxation processes in T1 such as the intersystem crossing from the triplet state T1 to the ground state S0, the reversible intersystem crossing between excited singlet S1 and triplet T1, as well as a rate of competiting intermolecular vibrational relaxation were compared. It was found that under vibrational equilibrium at high vibrational temperature (Tvib>>T) the experimentally observed nonexponential dependence of the gas phase triplet molecule decay time on Evib reflected the excess vibrational energy (in T1) dependence of intersystem crossing rate from T1 to S0 that was well approximated by Arrhenius’ activated type rate equation in contrast to exponential decay for an isolated molecule.
The photochemical and photophysical properties of carbonyl aromatic compounds in the triplet state are currently of interest due to their ability to participate in many important photochemical reactions sensitized by triplet ketones such as hydrogen atom, electron and proton transfer. Many aspects of these reactions are important for understanding of the fundamental processes in chemical and biological systems.
Much attention has been paid to decay processes in triplet states. In fact, great care was taken to study the sensitivity of triplet state decay time to both the intrinsic molecular parameters and environment properties in different media. Laser spectroscopy study of gas phase systems over a wide range of vibrational energies permitted us to obtain the important information about dominant pathways of triplet molecule relaxation. The additional data for the bath gas influence on the intensities and the decay rates of delayed fluorescence of vibrationally excited triplet molecules link spectroscopic information of an isolated molecule and a molecule in the dense medium. Gas phase data can be utilized to provide the prediction of the photophysics of complex molecules under study in a condensed phase over a broad temperature range.
In works [1,2], we demonstrated that delayed fluorescence (DF) induced by direct infrared multiphoton excitation (IRMPE) of triplet state molecules by CO2 laser radiation can be successfully used to study the excess energy dependence of nonradiative relaxation rates in the triplet state T1. It was noted that the advantage of this method was the possibility to change the vibrational energy of triplet molecules over a wide range because the intensity and decay rate of DF of vibrationally excited molecules proved to be very sensitive to a laser energy density.
In the present work, by utilizing the method for triplet molecule IRMPE we have studied the excess energy dependence for the intersystem crossing (ISC) rate from the triplet state T1 to the ground state S0, for the rates of the reversible ISC between the singlet state S1 and the triplet state T1 as well as the rates of competitive process of intermolecular vibrational relaxation in the state T1 within a wide vibrational energy interval. Such a complex study of nonradiative transitions permits the determination of the fate both of an isolated triplet molecule and a collisional perturbed one.
The chosen molecules of benzophenone (C13H10O), fluorenone (C13H8O) have IR absorption bands in the CO2 laser radiation region and can be IRMPE by CO2 laser radiation both in the ground electronic and in the triplet state T1. They process a high average vibrational energy áEvibñ = 3100 cm-1 at an initial temperature of 380K for benzophenone and áEvibñ = 3000 cm-1 at 428K for fluorenone and emit long–lived luminescence in the gas phase due to efficient singlet–triplet interaction between the first excited singlet S1 and triplet T1. At the initial temperature of experiment the delayed emissions mainly correspond to delayed fluorescence with a decay time of 160 ms for benzophenone (T=380K) and 420 ms for fluorenone (T=428K). It is worth noting, whereas benzophenone is characterized by a low electron energy gap, DEST @ 2000 cm-1, between S1 and T1, the corresponding value for fluorenone is much larger (5400<DEST<7000 cm-1).
The experimental technique used in the present work was described previously [1,2]. Rapid ISC of molecules under study was exploited to prepare them in the triplet state T1. The nitrogen laser was used to excite a molecule to the singlet state S1. IR excitation of triplet molecules was produced by TEA CO2 laser radiation that was focused by lenses from blenched Ge and was passed through a cell beam to a beam with visible light. The samples to be irradiated were prepared in a high vacuum line–provided cylindrical heated cell with NaCl windows. Zone refined substances were stored in the side arm of the quartz cell. A vapor pressure was controlled by a temperature of the thermostated side reservoir. Vapors were diluted with a large quantity of bath gases: He3, Kr, N2 (0<Pbg<20 Torr). Luminescence was observed in the direction normal to excitation. A signal of the photomultiplier placed behind the monochromator slit came to the digital oscilloscope and was averaged over several pulses by a computer.
For relaxation processes within a wide interval of the vibrational energy of the triplet molecules and bath gas pressure to be analyzed, the following results are of special importance.
1. CO2 laser activated emissions were observed only in the spectral regions of usual luminescence. The spectra were slightly shifted to the high–frequency region as compared to those of thermally activated molecules. This effect was caused by reducing of the role of “hot” phosphorescence and observed in other cases at nonequilibrium vibrational excitation of the triplet molecules.
2. As discussed in our previous works [1,2], the decay of CO2 laser induced DF proved to be nonexponential and exhibited two exponential decay behaviors characterized by two distinct decay rates:
I(n,t)=I1×exp(-K1t)+ I2×exp(-K2t)
where K1 and K2 were equal to 106, 104 s-1 and 103, 102 s-1, respectively. The decay rates of DF are independent of the observation wavelength for lob = 400–580 nm, as it is typical for DF of molecular systems characterized by the efficient ISC. The decay rates of a fast component of both benzophenone and fluorenone increased by several orders of magnitude with a rise of CO2 laser energy density ECO2. The dependence of K1 on ECO2 was essentially distinguishable at different bath gas pressure.
3. At energy densities ECO2 sufficient to induce DF of fluorenone the decay rates of the fast component of DF linearly increased with increasing bath gas pressure Pbg only in a limited range of pressures. At a higher pressure, the decay rates remained unaltered. The added bath gas caused the decrease of benzophenone DF decay rates K1 at low ECO2 and the increase at ECO2>1.2 J/cm2.
To consider photophysical processes in the state T1 at high Evib, let us analyze the features of intermolecular vibrational relaxation as a main competitive process after the MPE of triplet molecules. (1) An important feature of multiphoton excited molecules that have more than 60 vibrational degrees of freedom, is a rapid redistribution of excitation energy from the levels of excited vibration to the triplet manifold of vibrational states during an IR laser pulse. A time resolution of experiment permits us to study vibration–translation (V–T) relaxation and is insufficient to determine the decay rates of vibration–vibration (V–V) relaxation. (2) MPE of triplet molecules causes a population of quasicontinuum states. Therefore, the excitation process should be reliably considered to be incoherent. Thus, the time variation of vibrational level population may be obtained from solving a set of coupled rate equations for radiative pumping in a quasicontinuum. That is why, we can easily predict a vibrational level population and evaluate an average vibrational energy of triplet molecules. (3) In vapors at a pressure of 10-3<PV<0.15 Torr, when only several collisions may occur during a full IR laser pulse, we can neglect the influence of collisions on the population distribution. Maximum vibrational temperatures, Tvib, achieved at MPE were estimated from comparison of intensities and decay rates of laser activated and thermally activated DF. (4) Collisions between excited molecules and bath gas particles change the MPE conditions that now depend on ECO2 and Pbg. By varying these values, we can control the condition of establishing vibrational distribution during the laser pulse. As Pbg increases and ECO2 decreases, the population distribution over vibrational levels in the triplet states is shifted to the region of low energies. It is worth noting that at the achieved excitation rates, Kex, the bath gas pressures used were insufficient to establish the steady state regime (Kex=KV), at which Evib does not any more increase.
|
Table 1. Maximum values of Tvib at MPE of triplet molecules |
||||
|
E CO2 , J/cm2 |
Bath gas |
Pbg, Torr |
Tvib, K |
Evib, cm-1 |
Fluorenone |
||||
|
3.2 |
0 |
750 |
11386 |
|
|
3.75 |
800 |
12731 |
||
|
4.25 |
850 |
14123 |
||
|
5.00 |
950 |
17033 |
||
|
6.00 |
970 |
17633 |
||
|
6.00 |
N2 |
7 |
825 |
13421 |
|
10 |
730 |
10862 |
||
|
6.00 |
Kr |
7 |
860 |
14407 |
|
7.60 |
0 |
1000 |
18544 |
|
|
8.4 |
0 |
1040 |
19775 |
|
|
Kr |
7 |
940 |
16736 |
|
|
20 |
888 |
15211 |
||
Benzophenone |
||||
|
1.3 |
0 |
720 |
11315 |
|
|
He3 |
8 |
652 |
9422 |
|
|
22 |
598 |
8005 |
||
|
1.6 |
0 |
790 |
13368 |
|
|
2.3 |
0 |
916 |
17280 |
|
|
Kr |
4 |
784 |
13189 |
|
|
10 |
682 |
10244 |
||
|
He3 |
8 |
842 |
14953 |
|
|
18 |
767 |
12683 |
||
|
2.8 |
0 |
1010 |
20340 |
|
|
Kr |
12 |
850 |
15200 |
|
|
3.24 |
0 |
1100 |
23359 |
|
|
Kr |
4 |
1040 |
21338 |
|
|
19 |
947 |
18278 |
||
Intermolecular vibrational relaxation in mixtures of vibrationally excited molecules and bath gases reduces some part of the triplet molecules excited above the singlet state S1. As a result, the time–resolved intensities, I(n,t), of the fast component decrease and its decay rates K1 increase with Pbg. This experimental information was adopted to determine the quantitative characteristics of vibrational energy transfer such as a collisional efficiency b=Kcol/Z=DK1/DP×Z and average energy transferred per one collision áDEñ=Evib×b as well as their excess energy dependence. Here Kcol and Z are the rate constants of intermolecular vibrational relaxation and gas kinetic collisions, respectively. It was found that b and áDEñ have small values typical for the vibration–translation process. In mixtures with Kr, He3, N2 at vibrational energies used they change over the range 10-4<b<3×10-4 and 3<áDEñ<20 cm-1, thereby increasing with Evib in just the same way as the rate of intermolecular vibrational relaxation. Conversion of vibrational energy to translational one involves as much as 103–104 collisions.
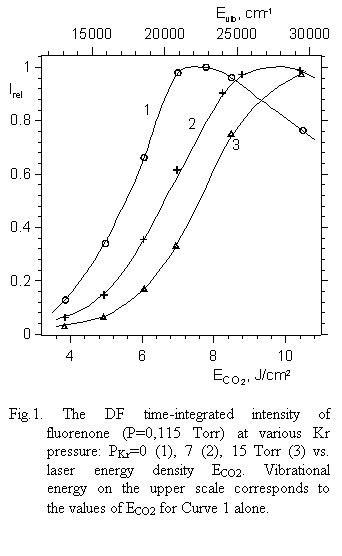 |
different ECO2 but at the same vibrational energy (Evib@22000 cm-1) in all cases.
|
Table 2. Integral intensities Iint and decay rates K2 of slow component for benzophenone DF at different Kr pressures PKr (Evib = 20340 cm-1) |
|||||||
|
PKr, Torr |
2 |
3 |
5 |
6 |
7 |
10 |
14 |
|
|
0.93 |
0.93 |
0.91 |
0.89 |
0.88 |
0.87 |
0.73 |
|
K2, 10-4c-1 |
6.5 |
6.5 |
6.4 |
6.2 |
6.0 |
6.0 |
5.8 |
The similar results are obtained for benzophenone. The dependences of K1 and I(n,t) on Pbg indicate that decreasing decay rates as well as leveling–off intensities take place at the same Evib@17000cm-1. At higher energies the time–integrated intensities and decay rates of fast and slow components of DF do not change mor
e with Pbg (Table 2). This is statistical limit behavior characterized by the absence of nonradiative relaxation rate dependence on pressure. Above the estimated Evib, the quasicontinuum conditions are fulfilled and DF decay is determined by intramolecular processes in T1. At vibrational energy under consideration, the collisional (Dncol@h×Kcol×P=10-6 cm-1) and laser broadening widths (Dnex@h×tP/sIL=10-4 cm-1) are much higher than a level energy difference because at this Evib we have a density of vibration levels as high as 1010 1/cm-1. Vibrational level densities higher than 106 1/cm-1 are necessary but insufficient to form quasicontinuum. It is good evidence that unharmonic coupling may exist only between specific modes of large complex molecules, but seems to be negligible for many other modes.
![Text Box:
Fig.2. Thr triplet decay rate plotted as a function of vibrational energy, Evib, in the triplet state. ▼, benzophenone at P=0.3 Torr (1); ►, benzophenone mixed with 11 Torr Xe (2); l, fluorenone at P=0.3 Torr; ♦, fluo-renone at P=0.04 Torr (4). Solid curves are the calculated values for Arrhenius rate equation. Open signs are gas-phase, solution and crystal lifetime results for benzophenone [3] and fluorenone [4] obtained by thermally activated DF and phosphorescence decay.](./content_files/image011.gif) |
The thermally averaged decay rate K(T) experimentally obtained under the conditions of high rates of intermolecular relaxation can be expressed in terms of a level density N(E) and microscopic decay rates of independently decaying levels, Kj, so that:
K(T) = òdENi(Ej)exp(-E/kT)Ki(Ej) / òdENi(Ej)exp(-E/kT). (1)
Thus, Eq. (1) couples microscopic time–resolved
spectroscopic information about an isolated molecule with kinetic information
in a dense medium and vice versa. In the present work, to predict the rates
of relaxation processes for an isolated molecule the solution was obtained for
the relevant set of coupled rate equations describing the population changes
in the states S0, S1 and T1. In order to avoid
unnecessary complications, the simplified kinetic scheme was used. The vibrational
levels of the triplet state were presented as a set of equidistant levels with
an energy gap of 0.1 cm-1. Only one level was suggested in S1.
Assuming also that the rates of reversible S1![]() T1 ISC are
determined by the density of vibrational levels in the final electronic states.
In addition, KTS0 was approximated by the relation: KTS0
= C1× exp(C2×Ej). In such a case, the necessary differential
equations can be written as:
T1 ISC are
determined by the density of vibrational levels in the final electronic states.
In addition, KTS0 was approximated by the relation: KTS0
= C1× exp(C2×Ej). In such a case, the necessary differential
equations can be written as:
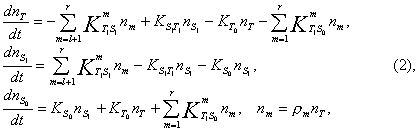
where nT = n0, nS1 = nS0 = 0 are the total populations in the states T1, S1 and S0 at t=0, nm is the population of the vibration level m, rm is the population distribution function satisfying the condition
![]()
l is the number of a vibrational level in the triplet state region where the triplet vibrational level does not yet interact with the vibrational level of the singlet state, i.e. DEST = 0.1(l+1) cm-1 (DEST @ 2000 cm-1 for benzophenone and DEST @6000 cm-1 for fluorenone were used). Temperature dependences of intensities and decay rates of thermally activated DF were employed to determine varying parameters l, C1 and C2. By utilizing the density of states calculated for benzophenone and for fluorenone and well-known radiative rates: KS0 = 3.3×105 с-1, KT0 = 1.4×102 с-1 for benzophenone and KS0 = 3×106 с-1, KT0 = 5 с-1 for fluorenone, we can predict absolute values of nonradiative process rates and their energy dependences.
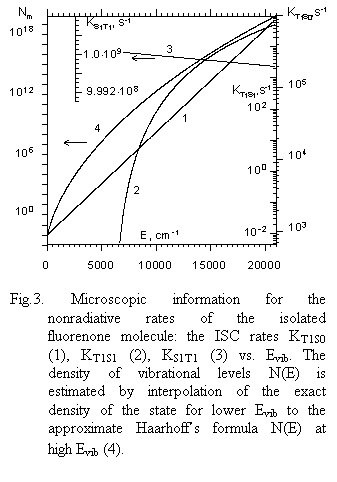 |
1) Nonradiative rate KT1S1 increases rapidly in an almost exponential fashion over a wide vibrational energy range.
2) KT1S0 appears to increase with vibrational energy of triplet molecules that is typical for radiationless transition rates.
3) KS1T1 decreases slowly with vibrational energy mainly due to a decrease of such a governing factor as the ratio NT(E)/NS(E) in a vibrational energy region under study. On the other hand, the coupling matrix element of singlet–triplet interaction is smeared out over a greater number of vibrational states, thus decreasing it as the density of vibrational states goes up with Evib. This collection of data was checked by utilizing the microscopic experimental data for decay rates of fluorenone DF obtained in a low pressure gas phase as well as by comparing the calculated DF pulse for benzophenone at given Evib with experimental one initiated by CO2 laser radiation. Experimental and calculated values prove to be in good agreement (Fig. 4).
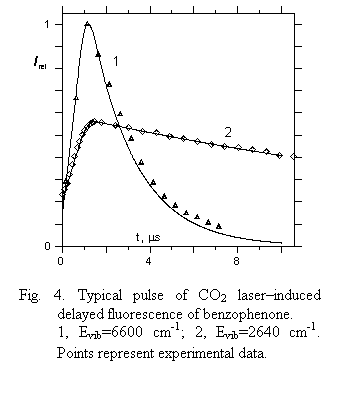 |
The energy dependence of nonradiative transition rates for the vibrationally excited triplet molecules was determined within the wide range of vibrational energies up to 20000 cm-1 (in T1). For gas phase benzophenone and fluorenone the rates of DF decay apparently varied by over several orders of magnitude as vibrational energy of the triplet molecules, Evib, ranged from 3000 cm-1 to 20000 cm-1.
V–T relaxation was found to be responsible for the pressure dependence of the DF decay rates after MPE of the triplet molecules by CO2 laser pulse (a half width of about 50 ns). The values of vibrational energies were estimated corresponding to the statistical limit conditions when the triplet molecule decay can be ascribed only by the unimolecular decay rates.
One
of the most interesting conclusion to emerge from this investigation was that
under vibrational equilibrium (KV> KTS0) at Tvib>>T
for gas phase benzophenone and fluorenone the ISC T1![]() S0 rate
dependences on Evib were well approximated by Arrhenius’ activated
type rate equation as well as it was found in many works for the temperature
dependence triplet molecule decay rates in condense neutral media.
S0 rate
dependences on Evib were well approximated by Arrhenius’ activated
type rate equation as well as it was found in many works for the temperature
dependence triplet molecule decay rates in condense neutral media.
Within the same vibrational energy range the energetic dependences of the rates KS1T1 and KT1S1 for reversible ISC between S1 and T1 demonstrated different behaviour for an isolated molecule: as the rate KT1S1 was an exponential function of Evib, whereas the rate KS1T1 decreased with Evib.
The results obtained may be important to understand the nature of triplet relaxation process and provide the prediction of the sensitivity of triplet molecule decay rates to intrinsic molecule parameters and ambient medium properties.
[1] N.A. Borisevich, G.A. Zalesskaya. Spectr. Lett., 19 (1986) 113–128
[2] G.A. Zalesskaya. J. Appl. Spectroscopy (ZhPS), 65 (1998) 657–693
[3] P.F. Jones, A.R. Calloway. Chem. Phys. Lett., 10 (1971) 438–443
[4] L.J. Andrews, A. Deroulede, H. Linschitz. J. Phys. Chem., 82 (1978) 2304–2309